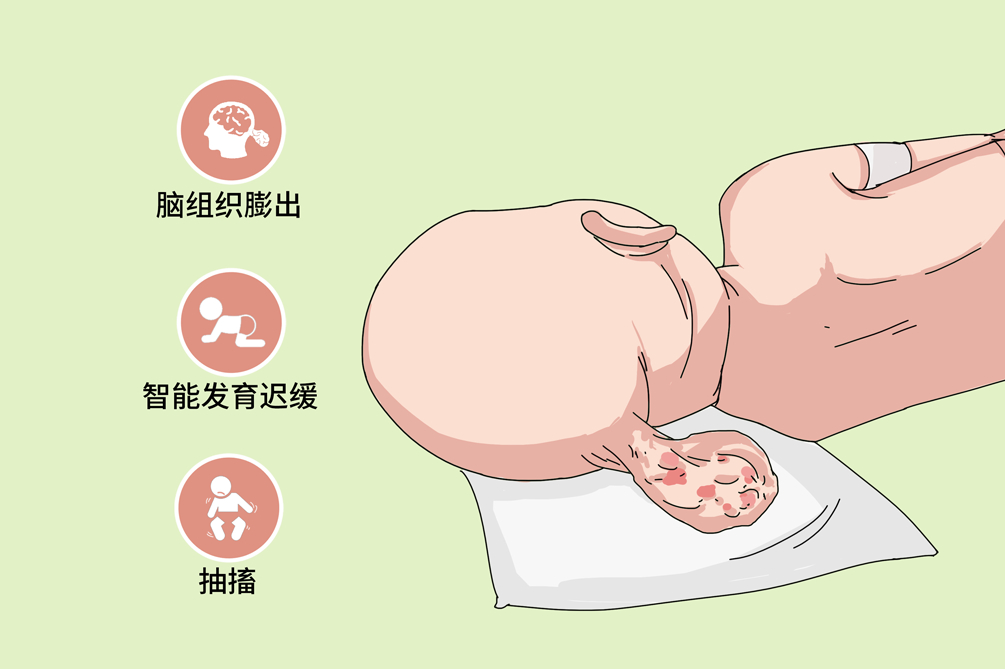
颅脑先天畸形是指由各种原因导致的先天性多种颅脑的形态改变和不同的神经系统功能障碍,包括先天性脑积水、颅裂、狭颅畸形等。临床较常见,大多数患儿由胚胎期神经系统发育异常所致,临床主要表现为脑组织外露、智能发育迟缓、抽搐等,部分需要手术治疗,预后因疾病而异。
颅脑先天畸形根据类型不同可分为以下几种:
由先天性因素引起的脑脊液分泌过多、循环受阻或吸收障碍而导致脑脊液在脑室和(或)蛛网膜下腔积聚,使脑室不断扩大、脑实质相应减少者,称为先天性脑积水。先天性脑积水习惯上主要分两大类,即交通性脑积水与阻塞性脑积水(也称非交通性脑积水)。另外还有脑外脑积水及多房性脑积水等比较特殊的脑积水。
颅裂为先天性颅骨闭合不全畸形,分为隐性及显性两大类。隐性颅裂少见,仅表现为颅骨缺损,而无软组织膨出;显性颅裂常同时存在脑膨出,包括脑膜膨出、脑膜脑膨出和积水性脑膜脑膨出。脑膨出好发于颅骨的中线部位,少数可偏于一侧,颅穹隆部、颅底部均可发生。发生于颅穹隆部者,可自枕、后囟、顶骨间、前囟、额骨间或颞部膨出;发生于颅底部者,可自鼻根部、鼻腔、鼻咽腔或眼眶等部膨出。以枕部和鼻根部脑膨出最为多见。
狭颅畸形又称颅缝早期闭合症或颅缝骨化症。在新生儿中的发病率为0.3%~0.5%。由于在生长发育过程中颅缝过早闭合,以致颅腔狭小不能适应脑的正常发育,可表现为颅内压升高、发育迟缓、智能低下、精神活动异常、癫痫发作等症状。治疗方法首选手术,多以整容为目的,一般于生后6个月至1岁时手术。
颅颈交界区由环绕枕骨大孔的枕骨、寰椎、枢椎及其附着的韧带围成的管状结构。颅颈交界区畸形是该区域颅骨、颈椎、脊髓、小脑及周围软组织发育异常形成的一组先天性疾病,主要包括颅底凹陷、扁平颅底和寰椎枕化、寰枢椎脱位、小脑扁桃体下疝和脊髓积水,严重患者常合并上述两种或多种畸形。
[1]王艳萍,梁娟,吴艳乔,朱军,缪蕾,周光萱,代礼.1988~1992年我国主要高危高发先天畸形发生率趋势分析[J].华西医学,2000,15(3):289-291.
[2]邢纯.超声对先天脑发育畸形的诊断[J].中外健康文摘,2012,(41):354-355.
[3]丁大明.颅骨先天畸形的影像诊断分析[J].中外健康文摘,2013,(4):171-172.
