黏多糖贮积症Ⅰ型是一组由溶酶体异常引起的遗传性黏多糖代谢障碍,由于酶的活性缺陷使不完全分解的氨基葡萄糖贮积而引起的先天性风湿病。黏多糖贮积症Ⅰ型的共同临床特征为有程度不等的骨骺改变、智力落后、内脏受累和角膜浑浊。生化特点是酸性黏多糖分解代谢缺陷,以致细胞内贮积过多的黏多糖,同时尿中也有过多的黏多糖排出。本病可通过手术治疗以及酶代替疗法改善,但预后较差。
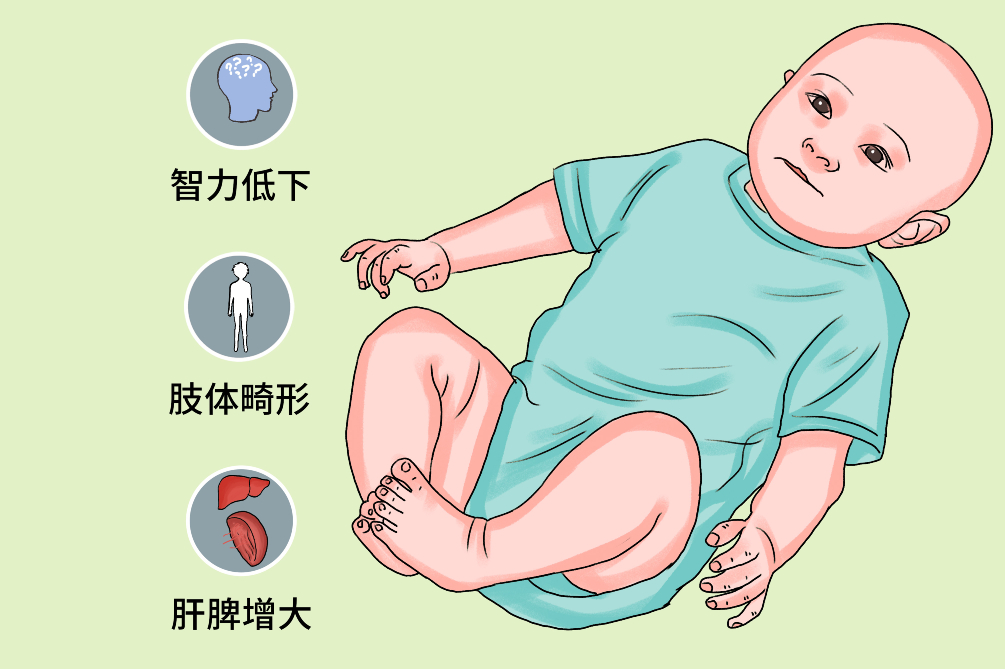
即MPS-IH型,为严重的黏多糖贮积症Ⅰ型,是最早被承认的经典型。α-L艾杜糖醛酸苷酶活性完全缺乏,使硫酸皮肤素和硫酸类肝素沉积于全身各组织。
即MPS-IS型,亦即7大类中原Ⅴ型(MPS-Ⅴ型),为黏多糖贮积症Ⅰ型中最轻的一型。酶缺乏与Hurler综合征相同,亦为α-L-艾杜糖醛酸苷酶缺乏。
本症极为罕见,亦为α-L-艾杜糖醛酸苷酶缺乏。
[1]唐正义.遗传与优生基础[M].西南师范大学出版社,2018.04:81.
[2]邬玲仟,梁德生.中华民族基因组多态现象研究人类单基因遗传疾病[M].西安交通大学出版社,2015.11:236.
[3]鲍秀兰.矮身材儿童266个怎么办[M].第2版.中国协和医科大学出版社,2015.03:163.
[4]程志伟,胡亚飞.实用医学影像学诊断[M].吉林大学出版社,2016.04:272.
[5]王金荣.实用儿科住院医师手册[M].山东科学技术出版社,2016.08:158.
