先天性外耳道闭锁

概述
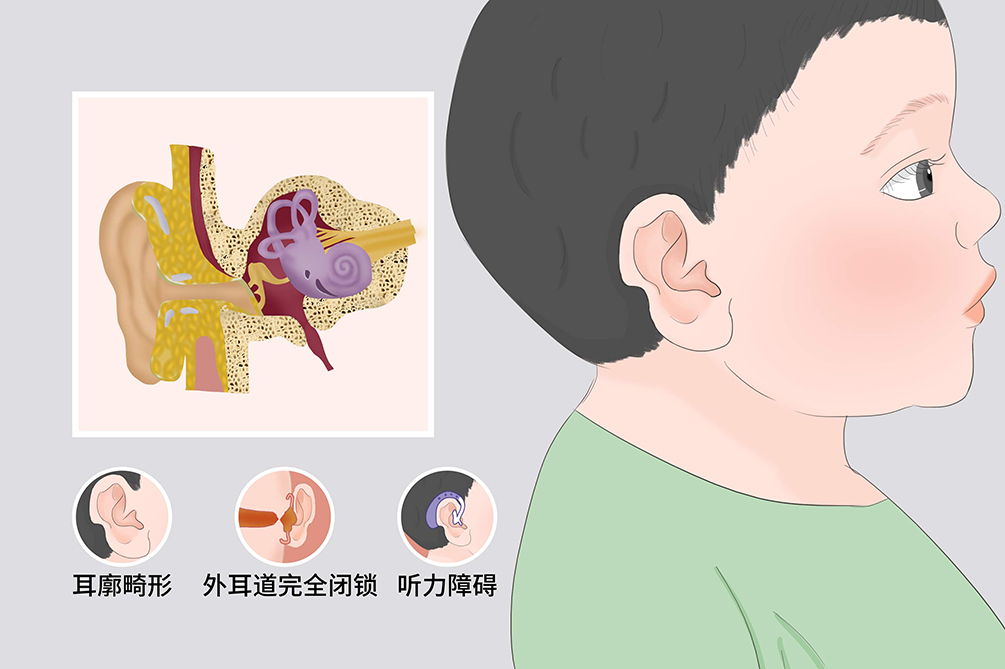
先天性外耳道闭锁是因胚胎期第一和第二鳃弓之间的第一鳃沟发育障碍所致,由于外耳道闭锁的先天畸形,引起单侧或双侧听力及语言障碍,一般内耳功能正常。治疗以手术为主,手术目的是提高听力与整形。双侧先天性耳畸形伴耳聋者应在4岁前进行手术,尽早提高听力。单侧先天性耳畸形者可稍缓手术。
- 就诊科室:
- 耳鼻喉科
- 是否医保:
- 是
- 英文名称:
- congenitalatresiaofexternalacousticmeatus
- 是否常见:
- 否
- 是否遗传:
- 是
- 并发疾病:
- 中耳畸形、先天性小耳、传导性耳聋
- 治疗周期:
- 短期手术治疗
- 临床症状:
- 无外耳道、听力障碍、语言学习障碍
- 好发人群:
- 直系亲属有先天性外耳道闭锁病史者、妊娠期间染病或用药不当者
- 鉴别诊断:
- 无
- 常用检查:
- 局部检查、听功能检查、影像学检查
疾病分类
由于外耳道发育不全常常合并中耳畸形,而且两者的畸形程度有一定的相关性,故目前有数种关于耳畸形的分型法,如Altmann(1955)分型法。
Ⅰ型(轻度):外耳道狭窄,鼓室发育不全,小鼓膜,鼓室正常或发育不全。
Ⅱ型(中度):外耳道闭锁,鼓室狭小,有闭锁板,听骨链畸形。
Ⅲ型(重度):外耳道闭锁,鼓室狭小或严重发育不全,听骨缺如或严重畸形。
4889点赞
参考文献
[1]陈艳丽.先天性外耳道闭锁的外科治疗[D].福建医科大学,2013.
[2]林海燕.耳鼻咽喉头颈外科临床护理路径[M].北京:中国医药科技出版社,2015.20.
[3]王玮,杜秀敏.当代实用医学概论下[M].哈尔滨:黑龙江人民出版社,2006.1046.
[4]周颖.儿童先天性外耳道闭锁成形术护理[J].护士进修杂志,2015,30(18):1673-1675.