Wolman病

概述
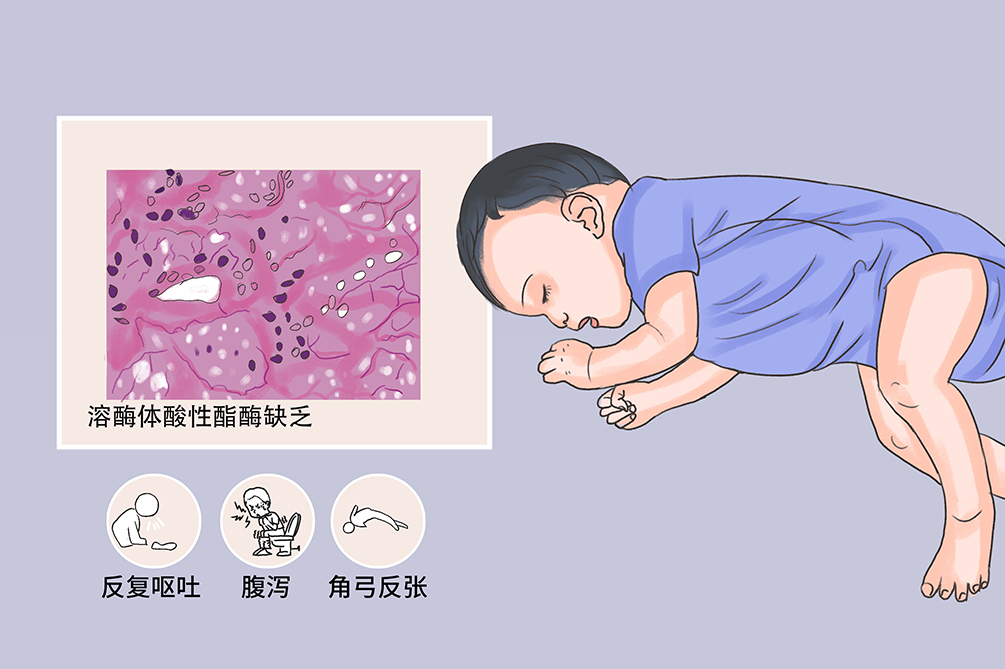
Wolman病为胆固醇水解酶的基因突变所致,主要是先天性溶酶体酸性酯酶缺乏,其特点是胆固醇酯和三酰甘油在体内蓄积,引起内脏器官黄瘤样改变,常累及肝、肾上腺、脾、淋巴结、骨髓、小肠、肺和胸腺等多个器官,是一种罕见的常染色体隐性遗传病,主要累及新生儿及儿童。病理上常出现大量酯化的胆固醇及三酰甘油在肝、脾、肾上腺中积聚。在1岁前发病,临床表现有喷射性呕吐、腹泻、腹部膨隆、肝脾肿大、黄疸、生长障碍、神经系统发育异常,表现为活动少、腱反射亢进、阵挛和角弓反张,以及肾上腺钙化。本病可使用造血干细胞移植和酶替代治疗等方法行病因治疗,同时可给予低脂饮食、肠外营养,以及糖皮质激素和盐皮质激素等对症治疗。Wolman病预后较差,轻症的胆固醇酯贮积病可不影响生存。
- 就诊科室:
- 儿科
- 是否医保:
- 否
- 英文名称:
- wolman disease
- 疾病别称:
- 沃尔曼病
- 是否常见:
- 否
- 是否遗传:
- 是
- 并发疾病:
- 贫血、消化道出血、败血症、昏迷
- 治疗周期:
- 短期治疗,1周~1月。
- 临床症状:
- 反复呕吐、腹泻、角弓反张
- 好发人群:
- 新生儿及婴幼儿,以及有家族遗传史者
- 常用药物:
- Sebelipase alfa、辛伐他汀、普伐他汀、依折麦布
- 常用检查:
- 血生化检查、溶酶体酸性脂肪酶活性检测、基因检测、干血纸片法、超声检查、肝组织病理学检查
参考文献
[1]胡锡琪,张文宏,朱虹光,张继明等.胡锡琪图解肝病 疑难肝病临床病理诊断与鉴别诊断[M].上海:上海科学技术出版社,2018,63.
[2]宋岐,马威,李沁园,刘敏.Sebelipase alfa-用于治疗溶酶体酸性脂肪酶缺乏症的药物[J].临床药物治疗杂志,2017,15(01):71-74.
[3]朱燕凤,张婷,陈扬,林凯,陆铸今,王晓红,俞蕙,陆国平.Wolman病临床及LIPA基因突变1例[J].中国循证儿科杂志,2013,8(01):55-59.