小儿糖原贮积病Ⅶ型

概述
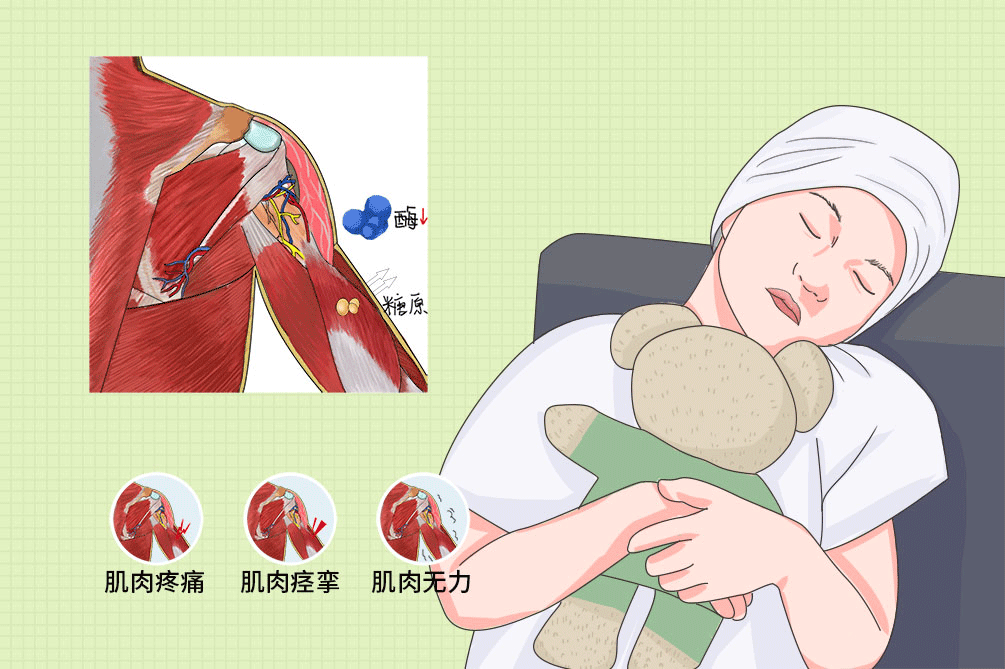
小儿糖原贮积病Ⅶ型是儿童的遗传性代谢疾病,主要是常染色体隐性遗传,是体内肌磷酸果糖激酶缺乏所引起糖原代谢性疾病。临床特征为肌肉无力,以体能活动能力降低和肌疼痛性痉挛为特征,休息或减慢活动速度即可使症状缓解。本病为遗传性疾病,不可治愈,仅需对症治疗为主。
- 就诊科室:
- 儿科
- 是否医保:
- 是
- 英文名称:
- Pediatric glycogen storage GSDS Ⅶ type
- 疾病别称:
- 肌磷酸果糖激酶缺乏症、小儿糖原累积病Ⅶ型
- 是否常见:
- 否
- 是否遗传:
- 是
- 并发疾病:
- 肌球蛋白尿症、高尿酸血症
- 治疗周期:
- 长期治疗
- 临床症状:
- 运动耐量降低、肌肉痉挛、肌肉无力
- 好发人群:
- 有糖原贮积病家族史的儿童
- 鉴别诊断:
- 炎症性肌病、酒精中毒性肌病、多发性肌炎
- 常用检查:
- 实验室检查、运动耐量检查、肌活检、肌电图检查、基因检测
疾病分类
根据疾病的发病年龄,可分为以下两类:
婴儿型
婴儿期即发病,患儿肌张力低下、肢体肌力差,表现为进行性肌病,常在4岁左右夭折。
成人型
以慢性进行性肌乏力为特征,肌肉痛性痉挛和肌红蛋白尿罕见。
参考文献
[1]王卫平,孙锟,常立文.儿科学(第9版)[M].北京:人民卫生出版社,2018:434-435.
[2]彭春梅,谢文光,何泳龙等.Ⅰ型和Ⅶ型糖原贮积病的致病基因及其作用机制研究进展[J].山东医药,2019,59(12):104-107.
[3]庄太凤,温春玲,王萍等.肌糖原累积症临床和遗传学特点研究进展.山东医药,2014,54(48):99-101.