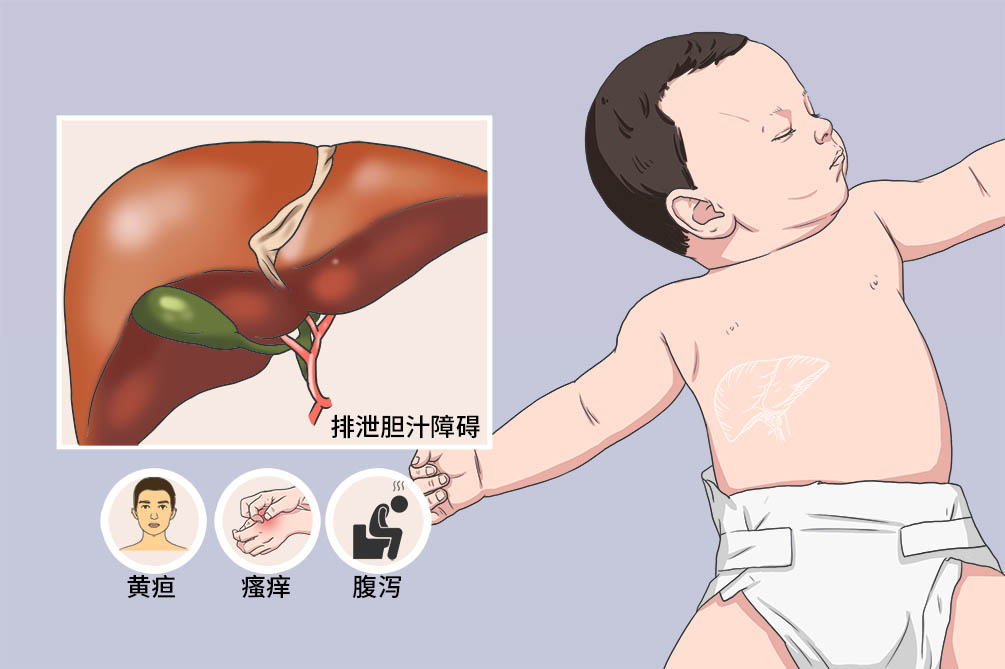
当进行性家族性肝内胆汁淤积症患儿出现黄疸和皮肤瘙痒时,应立即到儿童遗传代谢与内分泌科、儿童消化内科就医,可通过肝功能检查、病理检查、B超、基因检测确诊。但要与Crigler-Najjar综合征(先天性葡萄糖醛酸转移酶缺乏症)、Gilbert综合征(体质性肝功能不良性黄疸)、Lucey-Driscoll综合征(暂时性家族性高胆红素血症综合征)进行鉴别。
若患者出现严重的黄疸及瘙痒,并且有家族史时,优先考虑去儿童遗传代谢与内分泌科。
肝功能检查
进行性家族性肝内胆汁淤积症1、3型血清谷丙转氨酶轻度升高,2型中度升高。3型谷酰转肽酶有升高,1、2型均正常。2型甲胎蛋白有升高,1、3型均正常。1、2、3型结合胆红素和碱性磷酸酶都有升高。
病理学检查
1型的肝组织在电镜下可见颗粒状的胆汁,光镜下可见腺体胆汁淤积,轻度小叶纤维化。2型在电镜下可见无定型的胆汁,光镜下可见胆汁淤积、巨细胞肝炎、肝细胞坏死、门静脉纤维化。3型光镜下可见胆管增生,炎性浸润,胆道纤维化。
影像学检查
B超检查
一些进行性家族性肝内胆汁淤积症1型、2型患者可见胆囊内有结石。
磁共振胰胆管造影
有助于排除高胆汁淤积症患者的硬化性胆管炎和肝内外胆管结石。
基因检测
这是诊断该病的金标准,应用DNA测序检测致病基因外显子或者全基因测序。
患者有进行性家族性肝内胆汁淤积症家族史,表现为有严重黄疸、皮肤瘙痒、脂肪泻、生长发育迟缓等临床表现,全基因测序出现致病基因外显子可明确诊断。
Crigler-Najjar综合征(先天性葡萄糖醛酸转移酶缺乏症)
Crigler-Najjar综合征即先天性尿苷二磷酸葡萄糖醛酸转移酶缺乏。该病临床罕见,患儿很难存活,肝脏移植可以使尿苷二磷酸葡萄糖醛酸转移酶酶活性达到要求。本病主要是酶缺乏,而进行性家族性肝内胆汁淤积症主要由基因突变导致。
Gilbert综合征(体质性肝功能不良性黄疸)
Gilbert综合征是一种慢性的、良性高未结合胆红素血症,属常染色体显性遗传。Gilbert综合征症状轻,表现为皮肤巩膜黄染,通常于青春期才有表现。血液中未结合胆红素升高。进行性家族性肝内胆汁淤积症常呈现不同程度的黄疸,在婴儿期可能就有黄疸表现。
Lucey-Driscoll综合征(暂时性家族性高胆红素血症综合征)
Lucey-Driscoll综合征即家族性暂时性新生儿黄疸,本病有家族史,新生儿早期黄疸重,2~3周自然消退。该病可自然消退,预后较好,进行性家族性肝内胆汁淤积症一般不能自愈,需积极治疗。
胆道闭锁
先天性胆道闭锁或先天性胆总管囊肿,使肝内或肝外胆管阻塞,使结合胆红素排泄障碍,是新生儿期阻塞性黄疸的常见原因。B超、CT等影像学检查可见胆道闭锁,进行性家族性肝内胆汁淤积症一般无胆道闭锁,据此可鉴别两者。
新生儿肝炎
新生儿肝炎多由病毒引起的宫内感染所致,常在生后数天至数周内出现黄疸,持续时间较长,可伴有食欲下降、恶心、呕吐、消化不良、体重不增等症状。采血检查乙型肝炎病毒表面抗原,收集婴儿尿液或母宫颈刮片找巨细胞包涵体等可作病因诊断,由此与进行性家族性肝内胆汁淤积症鉴别。
Dubin-Johnson综合征(先天性结合型高胆红素血症)
Dubin-Johnson综合征即先天性非溶血性结合胆红素增高症,较少见。临床表现与进行性家族性肝内胆汁淤积症类似,以患者皮肤及巩膜黄染为主要症状,但症状一般较轻,呈间歇性反复发作。确诊需行染色体检查,由此与进行性家族性肝内胆汁淤积症鉴别。